This page was produced as an assignment for Genetics 677, an undergraduate course at UW - Madison.
The PAH Protein
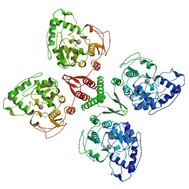
The structure of the PAH protein
The PAH protein is a 452 amino acid polypeptide that encodes phenylalanine hydroxylase. In cooperation with tetrahydrobiopterin (BH4), it is responsible for the conversion of phenylalanine to tyrosine by adding a hydroxyl group onto phenylalanine [1]. The protein exists as a homodimer and homotetramer, meaning that it is made up of two and four identical subunits, respectively. The protein also uses and binds Iron as a cofactor [2]. Each of the subunits contains areas that are responsible for catabolic activity and retaining the tetramer or dimer shape. Many of the mutations that cause PKU interfere with either of these structures [3]. Like many proteins involved in metabolism, the PAH gene is produced in the primarily in the liver, though it can also be found in minimal levels in the kidney and pancreas . Within the cell, the PAH protein functions in the cytosol, the part of the cytoplasm that is not taken up by organelles [2].
The PAH protein contains two domains: an ACT domain and a Biopterin_H domain. For more information regarding the functions of these domains, please visit the Motifs and Domains page.
One potential treatment for PKU is taking supplements of the chemical tetrahydrabiopterin (BH4). A synthetic version of this chemical, Kuvan, can be taken orally and has effect on about 10% of classic PKU cases. BH4 works by promoting function of any residual PAH activity that may be present [4].
The PAH protein is regulated in a couple different ways. There have been multiple studies that suggest global levels of Phe, along with phosphorylation of the protein are important in regulating protein activity. High levels of Phe tend to "open up" the catalytic section of the protein. Additionally, the binding of a phosphate group via cAMP onto the serine at residue 16 of the protein causes a conformational change that allows Phe access to the catalytic site [5,6].
UNIPROT Number: P00439 (PH4H_HUMAN)
The PAH protein contains two domains: an ACT domain and a Biopterin_H domain. For more information regarding the functions of these domains, please visit the Motifs and Domains page.
One potential treatment for PKU is taking supplements of the chemical tetrahydrabiopterin (BH4). A synthetic version of this chemical, Kuvan, can be taken orally and has effect on about 10% of classic PKU cases. BH4 works by promoting function of any residual PAH activity that may be present [4].
The PAH protein is regulated in a couple different ways. There have been multiple studies that suggest global levels of Phe, along with phosphorylation of the protein are important in regulating protein activity. High levels of Phe tend to "open up" the catalytic section of the protein. Additionally, the binding of a phosphate group via cAMP onto the serine at residue 16 of the protein causes a conformational change that allows Phe access to the catalytic site [5,6].
UNIPROT Number: P00439 (PH4H_HUMAN)
References
1. Mitchell JJ, Scriver CR. Phenylalanine Hydroxylase Deficiency. 2000 Jan 10 [updated 2010 May 04]. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available From http://www.ncbi.nlm.nih.gov/books/NBK1504/ PubMed PMID: 20301677.
2.UniProtKB. "Phenylalanine-4-hydroxylase - Homo Sapiens (Human)." N.p., 6 Feb. 2013. Web. 18 Feb. 2013. <http://www.uniprot.org/uniprot/P00439>.
3. Fusetti F, Erlandsen H, Flatmark T, Stevens RC. Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 1998 Jul 3;273(27):16962-7. PubMed PMID: 9642259.
4. KUVAN (sapropterin Dihydrochloride) Tablet." Daily Med. NCBI, Dec. 2007. Web. 07 May 2013.
5. McDowall, Jennifer. "Phenylalanine Hydroxylase." EMBL European Bioinformatics Institute.
6. Fitzpatrick PF. Allosteric regulation of phenylalanine hydroxylase. Arch Biochem Biophys. 2012 Mar 15;519(2):194-201. doi: 10.1016/j.abb.2011.09.012. Epub 2011 Oct 7. Review. PubMed PMID: 22005392; PubMed Central PMCID: PMC3271142.
2.UniProtKB. "Phenylalanine-4-hydroxylase - Homo Sapiens (Human)." N.p., 6 Feb. 2013. Web. 18 Feb. 2013. <http://www.uniprot.org/uniprot/P00439>.
3. Fusetti F, Erlandsen H, Flatmark T, Stevens RC. Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 1998 Jul 3;273(27):16962-7. PubMed PMID: 9642259.
4. KUVAN (sapropterin Dihydrochloride) Tablet." Daily Med. NCBI, Dec. 2007. Web. 07 May 2013.
5. McDowall, Jennifer. "Phenylalanine Hydroxylase." EMBL European Bioinformatics Institute.
6. Fitzpatrick PF. Allosteric regulation of phenylalanine hydroxylase. Arch Biochem Biophys. 2012 Mar 15;519(2):194-201. doi: 10.1016/j.abb.2011.09.012. Epub 2011 Oct 7. Review. PubMed PMID: 22005392; PubMed Central PMCID: PMC3271142.
