This page was produced as an assignment for Genetics 677, an undergraduate course at UW - Madison.
|
|
|
What is PKU?
Classic Phenylketonuria (PKU) is a rare genetic disorder that causes a complete or near complete inability to metabolize phenylalanine (Phe), an amino acid found in protein. If left untreated, PKU can lead to a number of medical conditions, such as significant and permanent mental retardation, learning disabilities, stunted growth, lighter-than-usual pigmentation of the skin and hair, behavior problems, seizures, and microcephaly (reduced head size) [1,2]. Fortunately, PKU is highly treatable, and by following a strict phenylalanine-free diet from infancy, a person born with PKU can grow up to lead a completely normal life.
According to the National PKU Alliance, approximately 14,500 people in the United States have PKU. This amounts to approximately 1/10,000 births, though incidence rates vary greatly between ethnic groups and country of origin. For example, people of Turkish origin experience an incidence of around 1/4000 births, while people of Finnish origin experience an incidence of approximately 1/100,000 births.
Note: There are other levels of severity of PKU (such as mild and moderate). Although they may have a degree of Phe intolerance, these forms of PKU are usually much easier to manage and are much less common. Due to the prevalence of Classic PKU and the severity in which the disorder presents itself, this website will concentrate on Classic PKU [1].
According to the National PKU Alliance, approximately 14,500 people in the United States have PKU. This amounts to approximately 1/10,000 births, though incidence rates vary greatly between ethnic groups and country of origin. For example, people of Turkish origin experience an incidence of around 1/4000 births, while people of Finnish origin experience an incidence of approximately 1/100,000 births.
Note: There are other levels of severity of PKU (such as mild and moderate). Although they may have a degree of Phe intolerance, these forms of PKU are usually much easier to manage and are much less common. Due to the prevalence of Classic PKU and the severity in which the disorder presents itself, this website will concentrate on Classic PKU [1].
The Science Behind It: The PAH Gene and Protein
PKU is an autosomal recessive disorder, meaning that a baby must inherit a copy of the defective gene from both parents in order to show symptoms of the disease. While not the exclusive cause of PKU, the gene most commonly involved is the pah gene, which is found on the long arm of chromosome 12 and encodes the protein phenylalanine hydroxylase (PAH protein) [3]. Currently, there are over 500 known mutations that can occur in the pah gene, many of which can render the gene defective [4]. Even if a child inherits one defective copy of pah, the remaining copy usually supplies a sufficient amount of phenylalanine hydroxylase for normal functioning. However, in the case of a child receiving two defective copies, the child will produce very little, if any, functional protein and therefore is incapable of breaking down Phe.
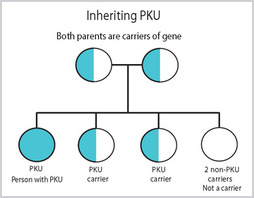
A sample pedigree for PKU, which is an autosomal recessive disorder.
|
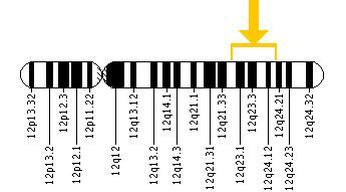
The location of the PAH gene on chromosome 12.
|
Usually, phenylalanine hydroxylase is used to convert phenylalanine into another amino acid, tyrosine. However, when there is no PAH, there is no conversion taking place, and Phe can build up in the blood. While someone with normal metabolic capabilities has a concentration of approximately 120 µmol/L of Phe in their blood, persons with untreated Classic PKU can have a concentration of over 1000 µmol/L [1]. This build-up of phenylalanine has a few implications, including the conversion of excess phenylalanine into phenylpyruvic acid and other by-products instead of tyrosine. This can cause the body to become starved for tyrosine, which is an important amino acid in many proteins [5]. Tyrosine deficiency has been associated with low levels of the neurotransmitter dopamine, which is important in executive functioning. Additionally, it is thought that the build-up of phenylalanine in the blood causes blockage of other similar amino acids (large neutral amino acids) from entering the brain. These other essential amino acids are important in the formation of neurotransmitters and other neural proteins such as myelin; the lack of access for brain cells to these amino acids causes a deficiency in these important proteins, which in turn can result in retardation [5,6].
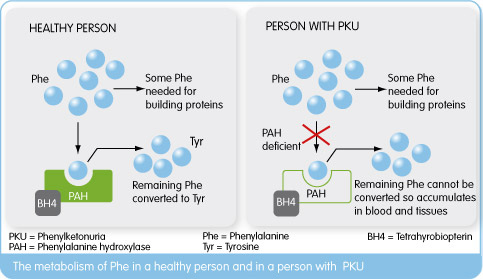
An illustration of Phe use in a healthy person vs. a person with PKU. BH4 is a necessary cofactor (i.e., helps PAH) in the synthesis of Tyrosine from Phenylalanine.
Treatment and Prevention

Fortunately, PKU is one genetic disorder that is almost completely controllable and treatable. Babies are tested within the first couple days of life with what is known as the "heel stick" test for an abnormally high amount of phenylalanine in their blood. Diagnoses can also come from urine tests, as the one of the by-products of phenylalanine, phenylketone (for which PKU is named), is present in urine if the child is afflicted [1].
Once a diagnosis is confirmed, the infant is put on a strict phenylalanine-free formula. When the child is old enough to eat other food, the diet is almost completely comprised of fruits and vegetables. Foods high in starch (such as pasta and potatoes), are very restricted but allowed in small amounts. Many people with PKU continue to take a form of special formula in order to get their required amount of protein and other nutrients that their diet lacks. All meats, dairy, nuts, and foods containing aspartame (which is made of phenylalanine and aspartic acid) are strictly prohibited in order to prevent toxic levels of phenylalanine build-up [1]. Close following of the diet can allow for completely normal development. It is also important that the diet remain continuous from infancy, as poor dietary habits can lead to lower IQ and issues with behavior, even if the diet is re-instated later on. However, research has shown that discontinuing the diet during adolescence only leads to "subtle" decreases in cognitive abilities compared to those who are lifelong dieters [6].
Additionally, taking a supplement of BH4, another substrate in the reaction leading to Phe degradation, has been shown to illicit a good response in managing about 10% of Classic PKU cases [7]. Theory also suggests that taking supplements of large neutral amino acids (which are important in brain development) could result in the "balancing" of the concentrations of these amino acids with the high concentration of phenylalanine in the blood. This could potentially allow more of these amino acids into the brain, reducing the risk of mental retardation [5,8].
Sticking closely to the PKU diet is especially important for women with PKU who are pregnant. Even if their unborn child has not inherited the disease, high levels of phenylalanine in the mother's blood can cause significant damage to the baby[1].
Once a diagnosis is confirmed, the infant is put on a strict phenylalanine-free formula. When the child is old enough to eat other food, the diet is almost completely comprised of fruits and vegetables. Foods high in starch (such as pasta and potatoes), are very restricted but allowed in small amounts. Many people with PKU continue to take a form of special formula in order to get their required amount of protein and other nutrients that their diet lacks. All meats, dairy, nuts, and foods containing aspartame (which is made of phenylalanine and aspartic acid) are strictly prohibited in order to prevent toxic levels of phenylalanine build-up [1]. Close following of the diet can allow for completely normal development. It is also important that the diet remain continuous from infancy, as poor dietary habits can lead to lower IQ and issues with behavior, even if the diet is re-instated later on. However, research has shown that discontinuing the diet during adolescence only leads to "subtle" decreases in cognitive abilities compared to those who are lifelong dieters [6].
Additionally, taking a supplement of BH4, another substrate in the reaction leading to Phe degradation, has been shown to illicit a good response in managing about 10% of Classic PKU cases [7]. Theory also suggests that taking supplements of large neutral amino acids (which are important in brain development) could result in the "balancing" of the concentrations of these amino acids with the high concentration of phenylalanine in the blood. This could potentially allow more of these amino acids into the brain, reducing the risk of mental retardation [5,8].
Sticking closely to the PKU diet is especially important for women with PKU who are pregnant. Even if their unborn child has not inherited the disease, high levels of phenylalanine in the mother's blood can cause significant damage to the baby[1].
References
1. Mitchell JJ, Scriver CR. Phenylalanine Hydroxylase Deficiency. 2000 Jan 10 [updated 2010 May 04]. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available From http://www.ncbi.nlm.nih.gov/books/NBK1504/ PubMed PMID: 20301677.
2. PubMed Health. (2011). Phenylketonuria. Retrieved January 27, 2013, from http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002150 [top]
3. "PAH." National Library of Medicine, Jan. 2008. Web. 30 Jan. 2013. <http://ghr.nlm.nih.gov/gene/PAH>.
4. PAH Locus Knowledgebase. N.p., 2009. Web. 30 Jan. 2013. <http://www.pahdb.mcgill.ca/cgi-bin/pahdb/mutation_statistics-1.cgi>.
5. van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only possible cause?J Inherit Metab Dis. 2009 Feb;32(1):46-51. doi: 10.1007/s10545-008-0946-2. Epub 2009 Jan 13. Review.
6. Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Arch Dis Child. 2007 Mar;92(3):213-8. Epub 2006 Oct 26. PubMed PMID: 17068073; PubMed Central PMCID: PMC2083434.
7. Blau N, Bélanger-Quintana A, Demirkol M, Feillet F, Giovannini M, MacDonald A, Trefz FK, van Spronsen FJ. Optimizing the use of sapropterin (BH(4)) in the management of phenylketonuria. Mol Genet Metab. 2009 Apr;96(4):158-63. doi: 10.1016/j.ymgme.2009.01.002. Epub 2009 Feb 8. Review. PubMed PMID: 19208488.
8. Schindeler S, Ghosh-Jerath S, Thompson S, Rocca A, Joy P, Kemp A, Rae C, Green K, Wilcken B, Christodoulou J. The effects of large neutral amino acid supplements in PKU: an MRS and neuropsychological study. Mol Genet Metab. 2007 May;91(1):48-54. Epub 2007 Mar 23. PubMed PMID: 17368065.
2. PubMed Health. (2011). Phenylketonuria. Retrieved January 27, 2013, from http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002150 [top]
3. "PAH." National Library of Medicine, Jan. 2008. Web. 30 Jan. 2013. <http://ghr.nlm.nih.gov/gene/PAH>.
4. PAH Locus Knowledgebase. N.p., 2009. Web. 30 Jan. 2013. <http://www.pahdb.mcgill.ca/cgi-bin/pahdb/mutation_statistics-1.cgi>.
5. van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only possible cause?J Inherit Metab Dis. 2009 Feb;32(1):46-51. doi: 10.1007/s10545-008-0946-2. Epub 2009 Jan 13. Review.
6. Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Arch Dis Child. 2007 Mar;92(3):213-8. Epub 2006 Oct 26. PubMed PMID: 17068073; PubMed Central PMCID: PMC2083434.
7. Blau N, Bélanger-Quintana A, Demirkol M, Feillet F, Giovannini M, MacDonald A, Trefz FK, van Spronsen FJ. Optimizing the use of sapropterin (BH(4)) in the management of phenylketonuria. Mol Genet Metab. 2009 Apr;96(4):158-63. doi: 10.1016/j.ymgme.2009.01.002. Epub 2009 Feb 8. Review. PubMed PMID: 19208488.
8. Schindeler S, Ghosh-Jerath S, Thompson S, Rocca A, Joy P, Kemp A, Rae C, Green K, Wilcken B, Christodoulou J. The effects of large neutral amino acid supplements in PKU: an MRS and neuropsychological study. Mol Genet Metab. 2007 May;91(1):48-54. Epub 2007 Mar 23. PubMed PMID: 17368065.
|
|
