This page was produced as an assignment for Genetics 677, an undergraduate course at UW - Madison.
What is Phylogeny?
Phylogeny refers to the evolutionary relationships and relatedness between different species or populations. Phylogenetic trees can be constructed on the basis of phenotypic relationships or on genetic relationships [1].
Phylogeny of the pah Gene
Once the homologs (and their sequences) are known, it is fairly easy to construct a phylogenetic tree. This involves inputting the sequences into a program like Clustal, which uses algorithms and statistics to predict evolutionary relationships between the homologs.
There are two different kinds trees that can be made: average distance or neighbor joining. Average distance trees use the alignments of homologous genes to infer closest relatives in context of the gene of interest. Neighbor joining trees, on the other hand, do not require alignments. Ultimately, neighbor joining trees are a form of an unrooted tree, that is, they show how many common ancestors are between two species, but do not infer how similar the sequences for a gene of interest are [2,3].
Additionally, each of these types of trees can be made using either % Identity or the BLOSUM62 matrix. Creating a tree with % Identity uses the actual sequences between homologs and how identical they are to create trees. BLOSUM62 references the likelihood of a certain amino acid being exchanged for another, and uses these likelihoods to determine how likely a certain mutation is bound to occur [4].
The problem with phylogeny lies in the fact that there are multiple algorithms that can be used. No algorithm is completely perfect, and as a result, depending on which algorithm is used, trees may look completely different from each other.
There are two different kinds trees that can be made: average distance or neighbor joining. Average distance trees use the alignments of homologous genes to infer closest relatives in context of the gene of interest. Neighbor joining trees, on the other hand, do not require alignments. Ultimately, neighbor joining trees are a form of an unrooted tree, that is, they show how many common ancestors are between two species, but do not infer how similar the sequences for a gene of interest are [2,3].
Additionally, each of these types of trees can be made using either % Identity or the BLOSUM62 matrix. Creating a tree with % Identity uses the actual sequences between homologs and how identical they are to create trees. BLOSUM62 references the likelihood of a certain amino acid being exchanged for another, and uses these likelihoods to determine how likely a certain mutation is bound to occur [4].
The problem with phylogeny lies in the fact that there are multiple algorithms that can be used. No algorithm is completely perfect, and as a result, depending on which algorithm is used, trees may look completely different from each other.
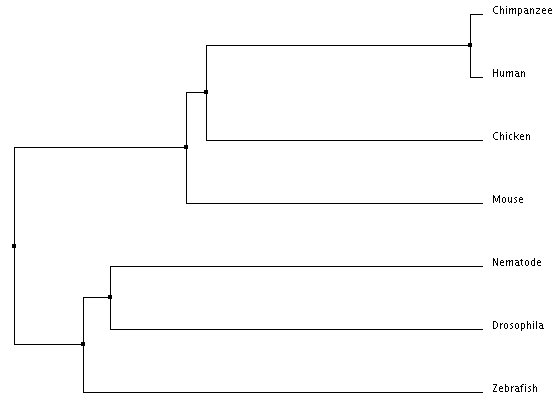
Average Distance Using Percent Identity (Click to Enlarge)
|
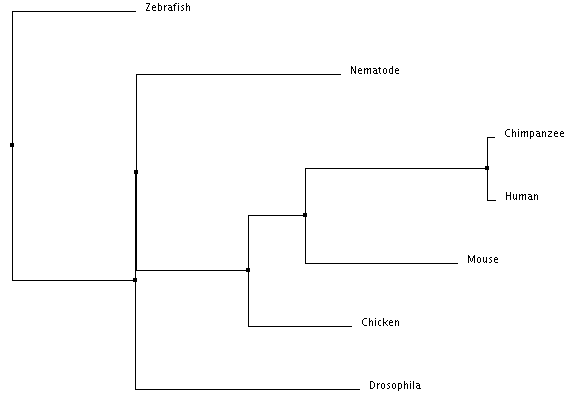
Neighbor Joining Using Percent Identity (Click to Enlarge)
|
Analysis and Discussion
The trees above were created using Clustal's Percent Identity algorithm. Both trees show that humans are most closely related to chimpanzees. As the homologies also show, chimpanzees and humans are more closely related to mice and chicken than they are to nematodes and fruit flies (Drosophila). However, there is a discrepancy between these two trees in regards to the mouse and chicken. The average distance tree suggests that the chicken is more similar to the chimpanzee and human than the mouse is. However, the neighbor joining tree places the mouse as more closely related to humans and chimpanzees than the chicken is. This latter result, from the neighbor joining tree, is in tandem with the homology results.
References
1. "Learning with the ToL." Tree of Life: What Is Phylogeny. Tree of Life Project, 2004. Web. 17 Feb. 2013. <http://tolweb.org/tree/learn/concepts/whatisphylogeny.html>.
2. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987 Jul;4(4):406-25. PubMed PMID: 3447015.
3. Opperdoes, Fred. "Neighbor-joining Method." Catholic University of Louvain, 8 Aug. 1997. Web. 18 Feb. 2013. <http://www.icp.ucl.ac.be/~opperd/private/neighbor.html>.
4. Staben, Chuck. "BLOSUM62 Matrix." BLOSUM62 Matrix. University of Kentucky, 28 Sept. 1998. Web. 17 Feb. 2013. <http://www.uky.edu/Classes/BIO/520/BIO520WWW/blosum62.htm>.
2. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987 Jul;4(4):406-25. PubMed PMID: 3447015.
3. Opperdoes, Fred. "Neighbor-joining Method." Catholic University of Louvain, 8 Aug. 1997. Web. 18 Feb. 2013. <http://www.icp.ucl.ac.be/~opperd/private/neighbor.html>.
4. Staben, Chuck. "BLOSUM62 Matrix." BLOSUM62 Matrix. University of Kentucky, 28 Sept. 1998. Web. 17 Feb. 2013. <http://www.uky.edu/Classes/BIO/520/BIO520WWW/blosum62.htm>.
